《基因療法》FDA要求臨床設計改為隨機、雙盲安慰劑對照試驗!! Taysha決定放棄治療巨軸突神經病變(GAN)的候選療法TSHA-120
秒速閱讀: 甚麼是FDA驗證的臨床試驗? 如果是FDA"同意"的臨床設計,為什麼"達標",但FDA卻不給核准? 這有迷思。其實很多生技公司都表示,只要沒安全性問題,公司有錢,基本上你就可以執行臨床試驗,但公司自己設的臨床試驗目標(解盲)過了,並不代表FDA就會通過你的藥證申請,以下就是很好的案例。還有更需注意,如果公司三期試驗的對照組是安慰劑組更需注意,除非是無藥可治(但FDA也可能要求現有維持治療方式的比較),不然市場已經一拖拉庫新藥或是學名藥,縱使你取得藥證可能競爭力有限,應該要與已經上市的"強者"頭對頭臨床試驗比,這樣未來上市的更具競爭力,也許就可搶食現有"強者"的市場。千萬不要為了"頭洗下去",就一定要完成沒有商業價值的臨床試驗,終究會傷了投資人的荷包。
Taysha是一家專注於開發和商業化基於AAV 的基因療法公司,在2023年9/19日表示,在收到與FDA C類會議後的回函-有關治療巨軸突神經病變 (GAN) 治療的 TSHA-120。
FDA C 類會議回函
FDA 繼續推薦隨機、雙盲、安慰劑對照試驗作為證明 TSHA-120 療效的最佳途徑。在其他方面,FDA 還提供了單臂試驗的潛在途徑,其中外部對照組與透過多種預後因素評估後進行治療的患者相比較,並建議進行長期追蹤以消除潛在的偏差。
根據美國 FDA 的 C 型會議回應,公司潛在生物製劑許可申請 (BLA) 所提交的三期研究設計之可行性面臨挑戰,Taysha決定停止在 GAN 中開發 TSHA-120。同時,公司將優先減少營運支出,並預計將現金使用延長至 2025 年第四季,以支持 TSHA-102 在雷特症候群評估中的持續開發。
Type-C 類會議
C 類會議基本上是一個「包羅萬象」的類別,包括任何涉及不屬於 A 類或 B 類範圍內的產品開發和審查的會議。 FDA 將嘗試在收到書面會議請求 75 天內試圖安排會議。當要求召開 C 類會議時,發起人(申請藥證者)可以要求對其問題進行書面答覆,而不是面對面會議、視訊會議或電話會議,但也可以要求舉行面對面會議,但 FDA 可能會決定只需要書面答覆。
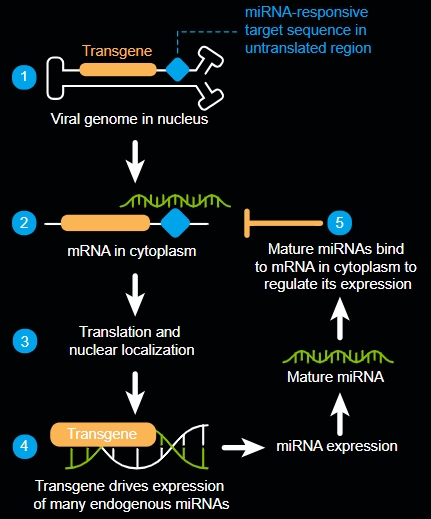
使用miRARE調節轉基因表達
在包括雷特氏症在內的許多疾病中,治療性基因轉殖的表達需要受到調節。高劑量的工程基因可能有害,而低劑量可能無效。對於需要替換劑量敏感基因的疾病,我們結合了高通量 microRNA (miRNA)、分析和基因組挖掘來創建 miRARE,這是我們的新型 miRNA 標靶組合。這種方法旨在使我們的候選產品能夠維持大腦中安全的轉基因表達水平。