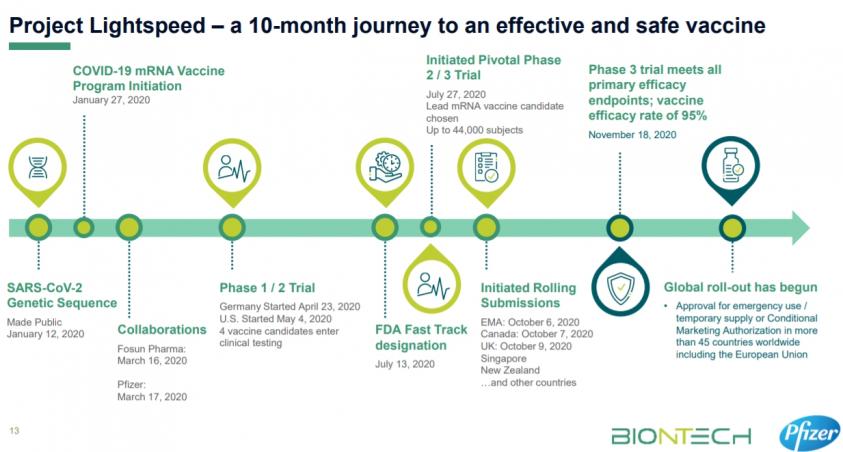
《FDA》申請緊急使用授權EUA 廠商需要提交三期臨床試驗的那些安全性和有效性的數據? 以輝瑞新冠疫苗為例
秒速閱讀:最近台灣最熱的疫苗話題就是,國產疫苗到底要不要進行三期臨床試驗? 無論如何,如果國產疫苗要走向國際取得國際認同的疫苗護照,全球性的三期似乎是不可免的。
也不少人文章認為輝瑞和Moderna可以取得FDA的緊急使用授權EUA是利用二期臨床試驗的數據,不過從輝瑞公告來看,FDA給與輝瑞EUA是基於包括在《新英格蘭醫學雜誌》上的關鍵三期臨床研究的數據-疫苗有效率為95%從第二次給藥後 7 天開始測量,同時不同年齡、性別、種族和民族人口統計數據的療效是一致的,所有試驗參與者將繼續接受監測,以評估長期保護和安全性。
對新冠肺炎疫苗臨床二期如何連結已取得EUA國際大廠的臨床三期的資料似乎還沒有一個定論,尤其現在又面對病毒不斷變種的病毒。至少我們可以參考被成為全球「黃金標準」的FDA核准緊急授權使用EUA的標準:
1. 新冠疫苗開發的標準非常嚴格,尤其涉及數萬名的受試者,未來更使用在數千萬甚至數億人或數十億人的身上。無論是臨床試驗的數據、真實世界的數據和製造數據都是疫苗開發公司必須全面提交給FDA進行審查評估。
2. 申請緊急使用授權EUA,疫苗開發公司可以提交三期臨床試驗的最終數據或是三期的期中分析數據(當初必須與FDA討論達到的主要觀察指標)給FDA進行審核,FDA再進行效益與風險的評估。
3. 提交將包括從使用疫苗進行的一期和二期研究中積累的所有安全性數據,預計三期數據將包括至少兩個月的中位隨訪,意味著在三期臨床試驗中,至少有一半的完成疫苗接種受試者在完成完整的疫苗接種方案後有至少兩個月的隨訪(可能一劑如嬌生或是輝瑞、AZ的兩劑)。
4. FDA也將對疫苗的化學、製造和品管數據的評估,以確保疫苗的質量和一致性。
相關閱讀: 【頭大大】壓力!? 國產疫苗三期臨床試驗最大挑戰,不是人數也不是疫區? 是一定要成功?
相關閱讀: 《FDA》緊急使用授權與完全核准差異在哪?輝瑞、Moderna力拼COVID-19疫苗的最後一哩路!(閱讀)
相關閱讀: 《COVID-19 疫苗》最後一哩路 ! 輝瑞/BioNTech 與摩德納(Moderna)為其新冠肺炎疫苗尋求正式批准 (閱讀)
|
以輝瑞BNT162b2的三期臨床申請EUA數據來說,
37,586 名參加正在進行的隨機、有安慰劑對照組的三期臨床試驗,在接受第二劑疫苗後的中位隨訪時間為兩個月。
現在關鍵三期期臨床試驗中的長期數據顯示,接種第二劑疫苗後的 6 個月,疫苗呈現出良好的功效與安全性。因此他們已向 FDA 滾動式地提交生物製劑許可申請 (BLA),以尋求全面批准這個用於 16 歲 (含) 以上的疫苗。 |